H.C.S
![]() Cette page est la première que je tiens à créer car notre petite famille est touchée par cette maladie.
Cette page est la première que je tiens à créer car notre petite famille est touchée par cette maladie.
Dans le cas de mon enfant, le dépistage a été fait à l'adolescence, suite à un arrêt de croissance. Le diagnostic a bouleversé de façon irréversible l'équilibre de toute notre maison, et nous a profondément meurtris...
Je n'oublierais jamais cette souffrance immense le jour de l'annonce qui a été faite à ma petite princesse, et le désarroi qui m'a saisi alors...Je pense à tous ces parents qui ont connu, ou qui connaissent la même douleur, et leur adresse tous mes voeux de courage et à leurs enfants, toute ma tendresse de mère..
 Dans les premiers jours qui suivent la naissance de votre enfant, à la maternité, il vous sera proposé de lui faire passer les tests dans le cadre de du programme national de dépistage.
Dans les premiers jours qui suivent la naissance de votre enfant, à la maternité, il vous sera proposé de lui faire passer les tests dans le cadre de du programme national de dépistage.
Ce dépistage permet de repérer les enfants atteints de certaines maladies graves, souvent d'origine génétique: La phénylcétonurie (PCU), L'hypothyroïdie congénitale, L'hyperplasie congénitale des surrénales, La mucoviscidose, La drépanocytose.
L'ensemble des tests est réalisé sur quelques gouttes de sang prélevées au talon de votre bébé, et recueillies sur une bandelette de papier buvard.
Les analyses sont ensuite effectuées par un centre de dépistage.
Ces analyses doivent parfois être complétées par une technique plus approfondie, dans ce cas, le centre de dépistage vous demandera votre accord par écrit.
Dans tous les cas, vous devez donnez votre accord oral pour la réalisation de cette analyse.
Si les résultats ne mettent pas en évidence l'une des 5 maladies dépistées, les analyses ne vous seront pas directement remises, mais elles seront à votre disposition à la maternité où le prélèvement a été effectué.
Si l'un des résultats montre la présence de l'une des 5 maladies, vous serez très rapidement informés dans les 15 jours qui suivent le prélèvement.
Pour la mucoviscidose, le résultat est un peu plus tardif, en raison du test génétique qui doit être effectué.
Hyperplasie congénitale des surrénales
L'HYPERPLASIE CONGENITALE DES SURRENALES est une pathologie secondaire à un déficit de la synthèse du cortisol dans la zone fasciculée de la glande corticosurrénale.
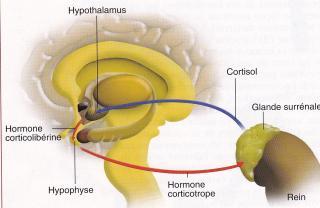
L'enzyme impliquée dans plus de 95 % des cas (la pathologie majoritaire donc) est la 21 -hydroxylase responsable de la transformation de la progestérone en désoxycorticostérone.
Il existe deux formes de déficit en 21 hydroxylase (ce fut notre cas) :
* Une forme classique avec un déficit enzymatique sévère et un début néonatal avec deux sous-types :
- une forme se manifestant par des signes de virilisation à la naissance (1/4 des cas)
- Une forme se manifestant par un syndrome de perte de sel avec risque vital dans les premières semaines de vie (3/4 des cas)
* Une forme non classique à début tardif et présenteront seulement des signes de virilisation à la puberté.
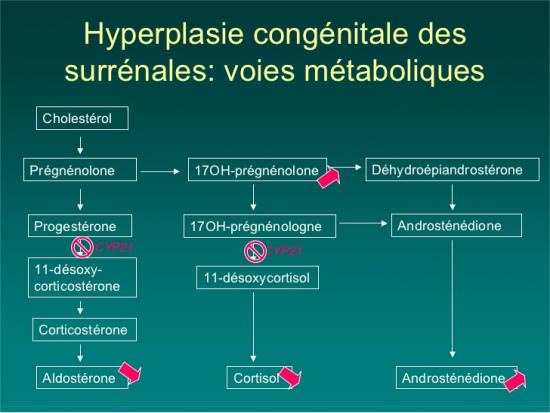
DEFICIT EN 21 HYDROXYLASE :
Mutation du gène CYP21A21
DEFICIT EN 3 BETA-HYDROXYSTEROIDE DESHYDROGENASE
Mutation du gène HSD3B2
DEFICIT EN 11 BETA-HYDROXYLASE
Mutation du gène CYP11B1
DEFICIT EN 17 ALPHA-HYDROXYLASE
Mutation du gène CYP17A1
FORME CLASSIQUE
- L'incidence est 1 sur 15 000 naissances en Europe, mais varie beaucoup en fonction de la population :
- 1 sur 300 chez certaines tribus esquimos
- 1 sur 5000 en Arabie saoudite
FORME NON CLASSIQUE
- 1 sur 40 chez les hispaniques
- 1 sur 50 chez les slaves
DESCRIPTION
DIFFÉRENCES CLINIQUES ENTRE LA FORME CLASSIQUE ET NON CLASSIQUE DE DÉFICIT EN 21 HYDROXYLASE Clinique Classique Non classique Virilisation prénatale Oui chez les filles Absent Virilisation postnatale Garçons et filles Variable Syndrome de perte de sel 3/4 des nouveau-nés Absent Déficit en cortisol Tous Rare
DIAGNOSTIC
Clinique
La suspicion congénitale des surrénales apparait lorsque :
Le nouveau-né féminin présente des signes de virilisation à la naissance, ou en raison de l'apparition de signes de virilisation (hirsutisme, découverte comme dans notre cas de taux de testostérone élevé...)ou présentant une puberté précoce (acné précoce, parfois comme pour nous dès la fin de maternelle..pilosité pelvienne précoce...)
Syndrome de perte de sel (forme grave) apparaissant dans les quatre premières années de la vie.
Il y avait avant le dépistage systématique des décés prématurés de petits garçons particulièrement, que l'on avait pas vu malades, et qui se déshydrataient lors d'une forte chaleur estivale, ou une forte fièvre...
Notre enfant est atteinte de la forme atténuée, dite non classique, de l'HCS, moins invalidante, car heureusement sans perte de sel.
BIOCHIMIE :
Dosage de la 17 OH progéstérone
Dosage de l'activité rénine plasmatique.
Génétique
L'étude ducaryotype doit confirmer la normalité de la formule chromosomique
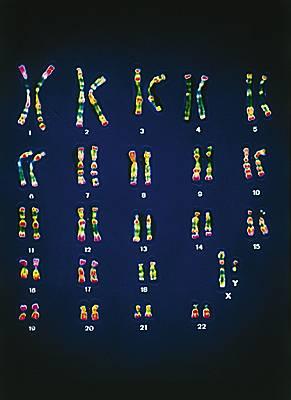
Exemple d'un caryotype masculin. Les deux chromosomes sexuels, un X et un Y, sont dissemblables.
TRAITEMENT
Le traitement dépend bien sûr des résultats biologiques et dosages pratiqués lors du bilan de la maladie.
Il s’agit d’un traitement substitutif à vie, de prise quotidienne. Il permet de supplémenter les hormones déficitaires et de freiner celles qui sont fabriquées en excès.
Le plus souvent on compensera le déficit en glucocorticoïde par de l'hydrocortisone.
Chez les sujets de sexe féminin, il convient de faire baisser le taux d'androgènes pour éviter la virilisation.
Par exemple on utilisera un dérivé de la progestérone avec action anti-androgène (Androcur par exemple).
MODE DE TRANSMISSION
Transmission autosomique récessive.
Explication : un caractère génétique est dit à transmission autosomique récessive quand :
1) le gène impliqué est porté par un autosome (chromosome non sexuel, ni X, ni Y, chez les organisme à systéme XY de détermination sexuelle);
2) Le phénotype associé de ce caractère est récessif (la présence de deux allèles identiques est indispensable pour que le caractère s'exprime.)
L'un des deux allèles est transmis par le gamète mâl, l'autre par le gamète femelle.
DEPISTAGE A LA NAISSANCE
Depuis 1996, le dépistage de l’hyperplasie congénitale des surrénales est devenu systématique, par dosage radio immunologique de la 17 hydroxyprogestérone sur le même papier buvard que pour les autres dépistages. Fait à 3 jours de vie, cela permet d’en faire le diagnostic très tôt, avant le syndrome de perte de sel qui survient habituellement vers 2-3 semaines de vie. Le nombre important de formes infra-cliniques, justifie ce diagnostic systématique.
![]()
Lien Association Surrénales http://www.surrenales.com
carte de soins et d'urgence insuffisant surrénalien http://surrenales.aphp.fr/fichiers/DGS-Insuff%20surrenale%20Soins%20et%20urgence.pdf
Informations ci-dessus trouvées sur http://www.afdphe.fr/

A COMMANDER EN CLIQUANT CI-DESSOUS :

En France, on compte aujourd'hui 7000 maladies rares et 3 millions de malades, soit une personne sur vingt atteinte !
La réalité de ces chiffres s'incarne dans des hommes, des femmes et des enfants, avec leurs difficultés pour vivre et leurs souffrances, et aussi, et surtout , la dignité et le courage dans la lutte contre la maladie.
Pour leur rendre hommage, Alliance Maladies Rares, collectif d'associations de malades, a décidé de donner la parole aux malades, aux familles et aux associations qui les soutiennent.
Des récits de vies, riches d'amour et de pudeur, émouvants de souffrances, surprenants d'humour parfois, et toujours nourris de révolte et d'espoir, décrivent les difficultés rencontrées et les combats menés lorsqu'on vit avec une maladie rare.
A COMMANDER EN CLIQUANT CI-DESSOUS :
Maladies rares : ils témoignent

A lire ce magnifique article sur le déficit en 21 hydroxylase
http://www.hebamme.ch/x_data/heft_pdf/2006-9-40.pdf
Ajouter un commentaire


